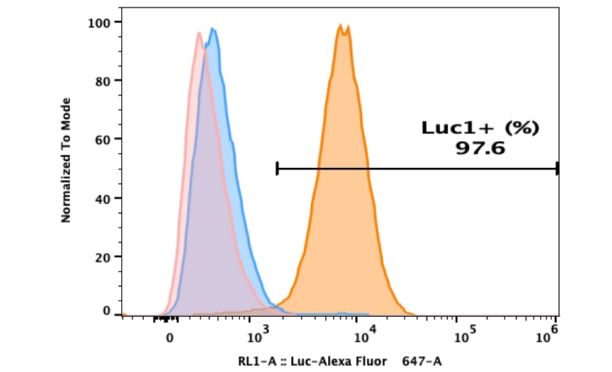
- In-Stock Tumor Cell Lines
- Human Orbital Fibroblasts
- Human Microglia
- Human Pulmonary Alveolar Epithelial Cells
- Human Colonic Fibroblasts
- Human Type II Alveolar Epithelial Cells
- Human Valvular Interstitial Cells
- Human Thyroid Epithelial Cells
- C57BL/6 Mouse Dermal Fibroblasts
- Human Alveolar Macrophages
- Human Dermal Fibroblasts, Adult
- Human Lung Fibroblasts, Adult
- Human Retinal Muller Cells
- Human Articular Chondrocytes
- Human Retinal Pigment Epithelial Cells
- Human Pancreatic Islets of Langerhans Cells
- Human Kidney Podocyte Cells
- Human Renal Proximal Tubule Cells
Knockin Stable Cell Lines
Knockin (KI) stable cell lines are genetically modified cell lines where a specific gene has been precisely inserted into a predetermined location within the genome. Unlike transient transfection where foreign DNA is only introduced into the cells temporarily, KI stable cell lines ensure consistent gene expression which is critical for studies requiring genetic fidelity [1]. KI stable cell lines also differ from knockout stable cell lines, where a gene is disabled, or overexpression stable cell lines, where a gene is introduced at a random location.
Creating KI stable cell lines has long been a tedious and unreliable process especially for researchers working on cancer therapy development or genetic disease studies. Compare to traditional methods which are time consuming (usually take a few months) and with low success rates, current advances in gene editing and delivery technologies are turning this bottleneck into a significant breakthrough.
Generation of KI stable cell lines
Limitations of Traditional Methods: Pitfalls of Random Integration
Conventional plasmid-based transfection frequently results in uncontrolled transgene copy numbers and positional effects, leading to highly variable expression levels. For example, GFP expression has been documented to vary over 100-fold in randomly integrated HEK293 lines due to differential chromatin context.
Recent Technological Advances
Significant improvements in the efficiency and precision of KI stable cell line generation have been driven by advances in gene editing and nucleic acid delivery systems. This section details the most promising contemporary approaches, their advantages over conventional methods, and essential experimental validation strategies.
1. CRISPR-Cas9 Mediated Homology-Directed Repair
1.1 Technological Principle
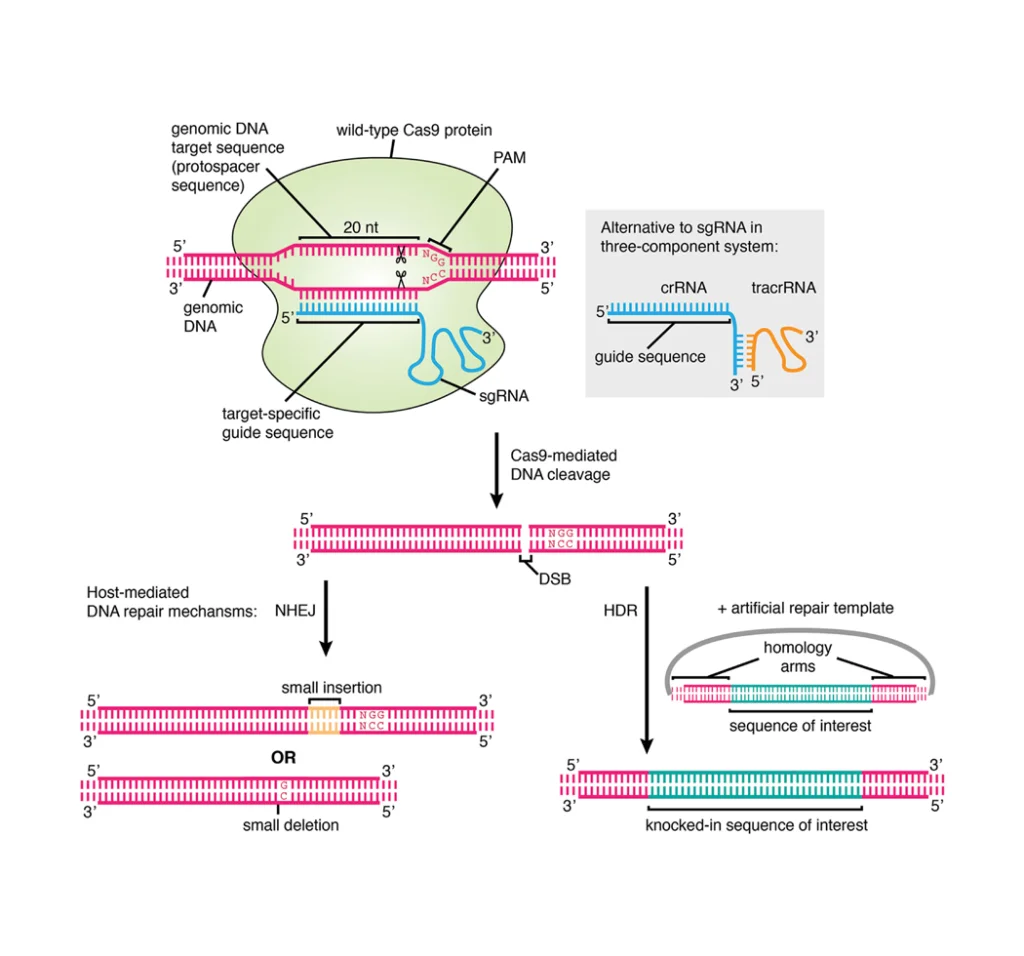
The CRISPR-Cas9 system facilitates precise gene knockin by inducing a targeted double-strand break (DSB) via the Cas9 endonuclease guided by a specific single-guide RNA (sgRNA). Precise integration of an exogenous DNA fragment into a predetermined genomic locus is achieved by co-delivering a homologous DNA repair template, which directs the cellular homology-directed repair (HDR) pathway.
Compared to the error-prone non-homologous end joining (NHEJ) pathway, HDR offers a high-fidelity mechanism essential for accurate gene knockin strategies and is primarily employed in CRISPR-Cas9-mediated gene knockin. Successful HDR-mediated integration necessitates the simultaneous introduction of the Cas9-sgRNA ribonucleoprotein (RNP) complex and a donor template possessing extended homology arms flanking the intended insertion site [2]. As shown in Figure 1, in the presence of a highly homologous DNA template, the HDR mechanism facilitates the integration of a DNA fragment into the target locus via homologous recombination. Such a repair mechanism allows for the precise insertion of a DNA sequence into a specific genomic location.
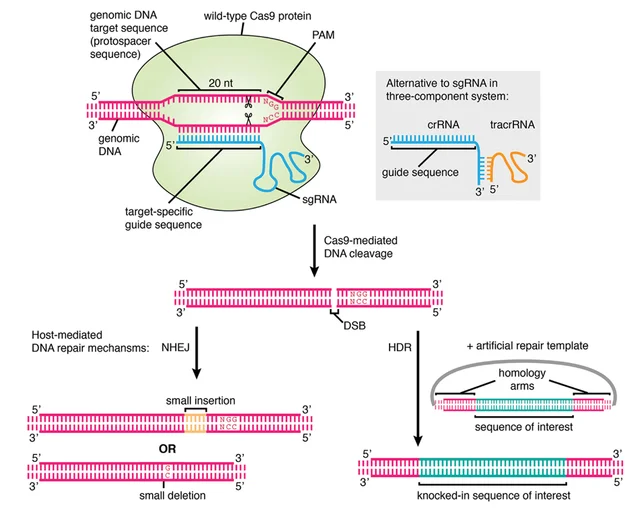
Figure 1. Schematic diagram of CRISPR-Cas9 based gene knockin.
2. Technical Process for KI Stable Cell Line Generation
2.1 Selection of Donor Template
Both double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA) are viable donor templates for HDR.
dsDNA templates are typically generated by PCR amplification or synthesis and contain the sequence of interest flanked by extensive homologous regions that can enhance HDR efficiency. This format is particularly suitable for the integration of large DNA fragments (Figure. 2) [3].
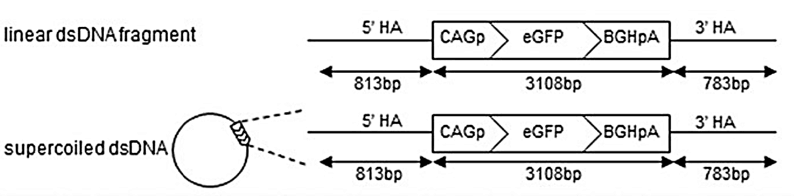
Figure 2. CRISPR-Cas9 mediated HDR strategy using dsDNA templates.
In contrast, ssDNA templates are single-stranded oligodeoxynucleotides (ssODNs) or synthesized ssDNA that are widely utilized due to their simpler structure, reduced probability of non-specific integration, and lower cytotoxicity. These properties enhance editing precision, making ssDNA advantageous for small insertions and point mutations (Figure. 3) [4].
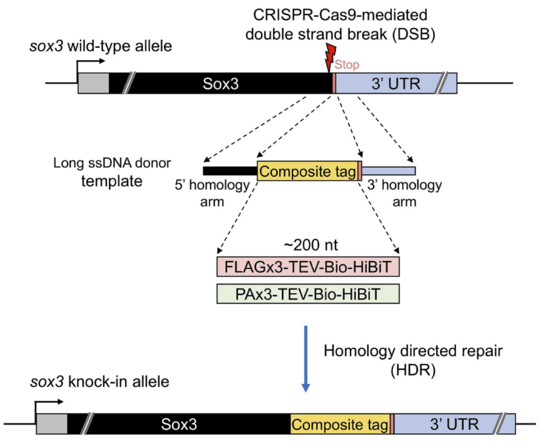
Figure 3. CRISPR-Cas9 mediated HDR strategy using ssDNA templates.
2.2 Design and Construction of sgRNA-Cas9 plasmids Componentsand repair DNA template plasmids Donor Template
2.2.1 Target Site Selection
① For non-codinginsertions, design sgRNA proximal to the insertion site. Construct the donor vector based on sequences flanking the sgRNA target region.
② For expression of non-fused proteins, targeting genomic safe harbor site (e.g., AAVS1, ROSA26) is recommended to minimize positional effects on expression.
③ For translational fusions, meticulous design of the linker peptide between the exogenous protein and the endogenous host protein is critical.
2.2.2 gRNA Design
Utilize established algorithms and online resources for optimal sgRNA design, such as:
CHOPCHOP: https://chopchop.cbu.uib.no/
2.2.3 sgRNA-Cas9 Plasmid/RNP Construction
Select the appropriate vector and construct the synthesized gRNA into the vector.
Clone the selected sgRNA sequence into an appropriate expression vector alongside Cas9 or assemble purified Cas9 protein with in vitro transcribed sgRNA to form RNP complexes.
2.2.4 Donor Plasmid/Template Design
① Homology Arm Design:
Design homology arms (~1000 bp) corresponding to the genomic sequences immediately upstream (Left Homology Arm, HA-L) and downstream (Right Homology Arm, HA-R) of the DSB site. Critically, the donor template must incorporate compensatory sequences corresponding to the insertion site. For instance, inserting EGFP immediately after the stop codon of Gene A requires incorporating the corresponding nucleotide sequence encoding the amino acids between the sgRNA cut site (e.g., 10 aa upstream) and the stop codon into the HA-L.
② Donor Vector Assembly:
Clone the gene of interest (GOI) between the HA-L and HA-R sequences. This complete cassette (HA-L-GOI-HA-R) is then subcloned into a suitable plasmid backbone for delivery.
2.3 Selection of Delivery Method
As indicated in the flowchart in Figure 4, the choices of delivery methods usually include Adeno-Associated Virus (AAV), RNP and plasmid methods. In Table 1, we have summarized the impacts of selection of different delivery methods in efficiency, safety and applicability.
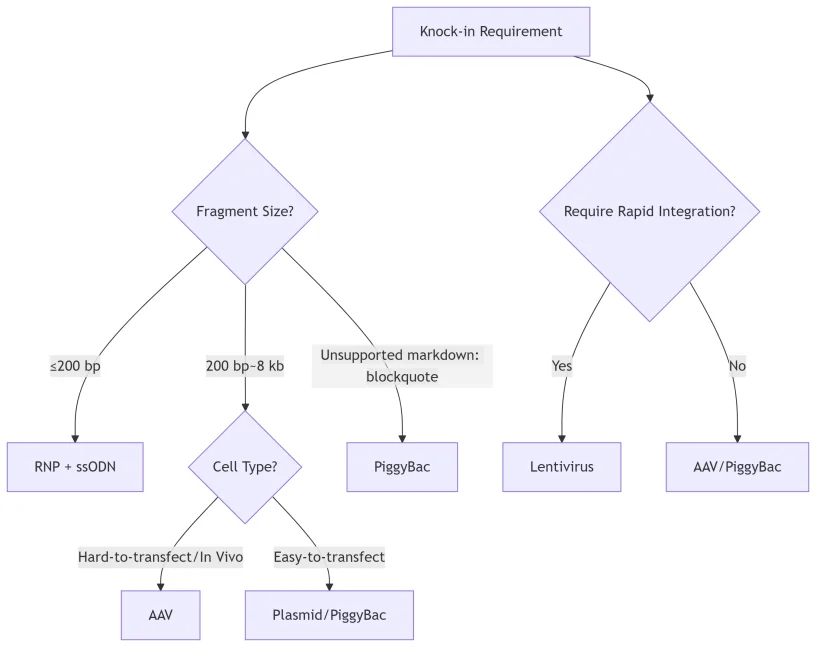
Figure 4. Flowchart for the selection of KI delivery methods.
Selection suggestions:
- High-Precision Small Fragment Editing (e.g., point mutations):RNP complexes + ssODN donors.
- Large Fragment Delivery In Vivo: AAV.
- Cost-Effective Screening: PlasmidDNA.
- Stable Integration of Large Constructs (e.g., antibodies, reporters, circuits): PiggyBactransposon system.
- Rapid Integration (e.g., CAR-T engineering, in vitro screening): Lentiviral Vectors (LV).
Table 1. A comparison of different delivery methods.
| Method | Efficient | Applicable cells | Carrier capacity | Off-target risk |
|---|---|---|---|---|
| AAV | Medium | Hard-to-transfect cells [5] | < 4.7 kb | Low |
| RNP | High | Primary cells including immune cells [6] | < 2 kb | Very low |
| Plasmid | Low | Easily transfected cell lines [7] | > 10 kb | Medium |
| LV | Medium | Actively dividing cells (such as T cells and hematopoietic stem cells) [8] | ≤ 8 kb | High |
| PiggyBac | Relatively high | A variety of mammalian cells (such as HEK293, iPSCs, and T cells) [9] | ~100 kb | Relatively low |
2.4 (Optional) Preliminary verification
Conduct preliminary screening using fluorescent marker proteins co-expressed with the sgRNA or via antibiotic resistance genes present in the donor or Cas9/sgRNA vectors.
2.5 Genomic PCR Verification
Design a pair of PCR primers annealing to the genomic region upstream of the 5′ homology arm and downstream of the 3′ homology arm of the targeted gene. Successful knockin is indicated by a increase of PCR product size corresponding to the inserted fragment compared to the wild-type allele.
2.6 Monoclonal Cell Line Isolation
① Fluorescent Reporter-Based Screening:
Utilize Fluorescence-Activated Cell Sorting (FACS) to analyze and isolate single cells expressing reporters (e.g., GFP/RFP).
② Antibiotic Resistance Selection :
Apply selective pressure (e.g., puromycin, hygromycin, or G418) followed by limiting dilution cloning to derive monoclonal populations.
③ Surface Marker-Based Screening:
For non-reporter or non-resistant cells, employ antibody staining against specific surface markers introduced by the KI, followed by FACS isolation.
2.7 MolecularValidation
Employ the verification methods such as Sanger sequencing to confirm the correct integration and sequence integrity across the insertion junctions, and verify the protein expression level and size via Western Blot (WB) analysis.
2.8 Expansion and Cryopreservation
Expand the validated monoclonal KI stable cell lines and prepare cryopreserved stocks for long-term storage and future experiments.
Applications of KI Stable cell lines
1 Drug Discovery & Screening
KI stable cell lines engineered to harbor specific disease-associated mutations (e.g., APP mutations for Alzheimer’s disease) provide genetically accurate platforms for high-throughput screening. For instance, isogenic Presenilin 1 (PSEN1) KI induced pluripotent stem cells (iPSCs) revealed subtle effects of γ-secretase modulators that were not discernible in conventional overexpression models [10].
2 Disease Mechanism Studies
Endogenous gene KI models recapitulate patient genetics with superior fidelity compared to cDNA overexpression. A 2022 study utilized BRCA1 KI stable cells to elucidate the functional consequences of splice-site mutations on protein activity in ovarian cancer pathogenesis [11].
3 Cell Therapy Development
CAR-T therapies critically depend on KI strategies to integrate the chimeric antigen receptor transgene into defined genomic safe harbor loci (e.g., TRAC locus). This targeted approach mitigates the risks of variegated expression and transcriptional silencing inherent to viral vector-mediated random integration [2].
Conclusion
Gene knockin techniques which involve inserting a specific DNA sequence into a targeted location in the genome, are generally more challenging than gene knockout techniques, where a gene is inactivated. So far, AcceGen has successfully offered more than 110 engineered KI stable cell lines with permanent knockin of specific GOI at desired genomic target sites via CRISPR-based approaches. Our proprietary technologies allow us to achieve very efficient gene delivery and HDR, leading to high successful rate and rapid turnaround for obtaining pooled or single clone deliverables.
References
[1] T. Gaj, C.A. Gersbach, C.F. Barbas, 3rd, ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering, Trends in biotechnology 31(7) (2013) 397-405.
[2] A. Agrotis, R. Ketteler, A new age in functional genomics using CRISPR/Cas9 in arrayed library screening, Frontiers in genetics 6 (2015) 300.
[3] S. Okamoto, Y. Amaishi, I. Maki, T. Enoki, J. Mineno, Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs, Scientific reports 9(1) (2019) 4811.
[4] D.C. Ranawakage, K. Okada, K. Sugio, Y. Kawaguchi, Y. Kuninobu-Bonkohara, T. Takada, Y. Kamachi, Efficient CRISPR-Cas9-Mediated Knock-In of Composite Tags in Zebrafish Using Long ssDNA as a Donor, Frontiers in cell and developmental biology 8 (2020) 598634.
[5] I.A. Anzai, L. Shaket, O. Adesina, M. Baym, B. Barstow, Rapid curation of gene disruption collections using Knockout Sudoku, Nature protocols 12(10) (2017) 2110-2137.
[6] S. Kim, D. Kim, S.W. Cho, J. Kim, J.S. Kim, Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins, Genome research 24(6) (2014) 1012-9.
[7] H. Yang, H. Wang, C.S. Shivalila, A.W. Cheng, L. Shi, R. Jaenisch, One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering, Cell 154(6) (2013) 1370-9.
[8] J.G. Doench, E. Hartenian, D.B. Graham, Z. Tothova, M. Hegde, I. Smith, M. Sullender, B.L. Ebert, R.J. Xavier, D.E. Root, Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation, Nature biotechnology 32(12) (2014) 1262-7.
[9] W. Kim, T. Gilet, J.W. Bush, Optimal concentrations in nectar feeding, Proceedings of the National Academy of Sciences of the United States of America 108(40) (2011) 16618-21.
[10] T. Kondo, M. Asai, K. Tsukita, Y. Kutoku, Y. Ohsawa, Y. Sunada, K. Imamura, N. Egawa, N. Yahata, K. Okita, K. Takahashi, I. Asaka, T. Aoi, A. Watanabe, K. Watanabe, C. Kadoya, R. Nakano, D. Watanabe, K. Maruyama, O. Hori, S. Hibino, T. Choshi, T. Nakahata, H. Hioki, T. Kaneko, M. Naitoh, K. Yoshikawa, S. Yamawaki, S. Suzuki, R. Hata, S. Ueno, T. Seki, K. Kobayashi, T. Toda, K. Murakami, K. Irie, W.L. Klein, H. Mori, T. Asada, R. Takahashi, N. Iwata, S. Yamanaka, H. Inoue, Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness, Cell stem cell 12(4) (2013) 487-96.
[11] S. Stella, S.R. Vitale, F. Martorana, M. Massimino, G. Pavone, K. Lanzafame, S. Bianca, C. Barone, C. Gorgone, M. Fichera, L. Manzella, Mutational Analysis of BRCA1 and BRCA2 Genes in Breast Cancer Patients from Eastern Sicily, Cancer management and research 14 (2022) 1341-1352.
Copyright - Unless otherwise stated all contents of this website are AcceGen™ All Rights Reserved – Full details of the use of materials on this site please refer to AcceGen Editorial Policy – Guest Posts are welcome, by submitting a guest post to AcceGen you are agree to the AcceGen Guest Post Agreement – Any concerns please contact marketing@accegen.com