- In-Stock Tumor Cell Lines
- Human Orbital Fibroblasts
- Human Microglia
- Human Pulmonary Alveolar Epithelial Cells
- Human Colonic Fibroblasts
- Human Type II Alveolar Epithelial Cells
- Human Valvular Interstitial Cells
- Human Thyroid Epithelial Cells
- C57BL/6 Mouse Dermal Fibroblasts
- Human Alveolar Macrophages
- Human Dermal Fibroblasts, Adult
- Human Lung Fibroblasts, Adult
- Human Retinal Muller Cells
- Human Articular Chondrocytes
- Human Retinal Pigment Epithelial Cells
- Human Pancreatic Islets of Langerhans Cells
- Human Kidney Podocyte Cells
- Human Renal Proximal Tubule Cells
Diabetes Mellitus
Diabetes mellitus (DM), generally known as diabetes, is a harmful condition characterized by persistently elevated blood glucose levels due to inadequate insulin produced by the pancreas or when the body fails to responding to the effects of insulin. By April 2025, diabetes affects nearly 589 million people globally, meaning one in nine adults are now living with the disease. Diabetes affects people of all ages. The estimated number of adults with diabetes is predicted to reach 853 million by 2050.
Diabetes, hyperglycemia (high blood glucose levels), and hypoxia (low oxygen levels in tissues) are interconnected in the context of diabetes complications. Hyperglycemia, a hallmark of diabetes, impairs the body’s ability to respond to hypoxia by destabilizing hypoxia inducible factor 1 alpha (HIF-1α), a protein crucial for adaptation to low oxygen conditions. Figure 1 illustrates the multifaceted mechanisms by which hyperglycemia disrupts HIF-1α stability and function in diabetes [1]. Such an interplay exacerbates the damage caused by both hyperglycemia and hypoxia, contributing to various diabetic complications including diabetic nephropathy, retinopathy, neuropathy, and cardiovascular disease.
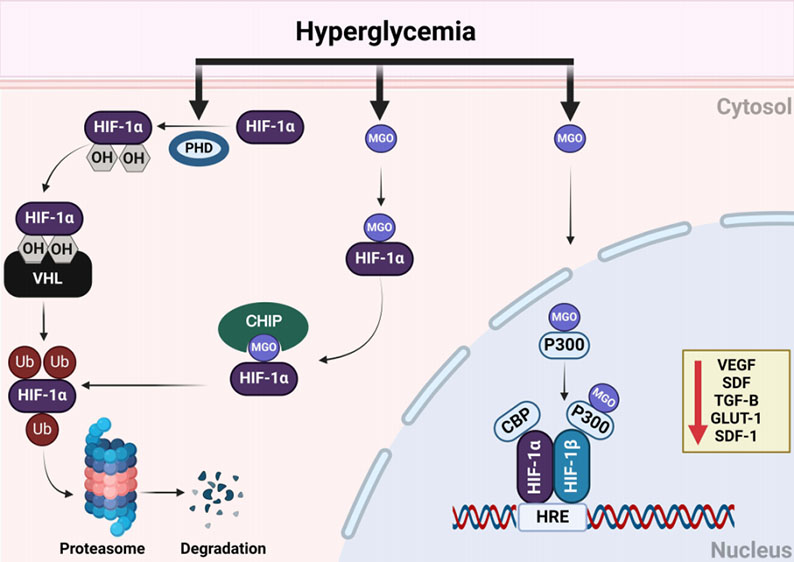
Figure 1. Hyperglycemia induced destabilization and dysfunction of HIF-1 signaling in diabetes.
Diabetic Kidney Disease
Diabetic nephropathy, also known as diabetic kidney disease (DKD), is a progressive kidney disorder that results from the damage caused by high blood glucose levels. This serious complication of diabetes significantly contributes to illness, disability, and healthcare costs. Diabetes is the leading cause of kidney disease, and up to 40% of adults with diabetes also have DKD. DKD is also the leading cause of end-stage renal disease (ESRD) in many developed countries including the United States [2]. Actually, excess mortality associated with type 1 and type 2 diabetes is largely confined to those with DKD. Consequently, preventing and managing DKD in patients with diabetes is a key aim of the overall management [3].
Traditionally, markers of DKD include albuminuria and creatinine, as well as estimated glomerular filtration rate (eGFR). However, these detection are always delayed and have limited utility. To fully elucidate the underlying mechanism and progression of DKD, researchers ought to focus more on the primary pathological features of DKD such as glomerular hypertrophy, dysfunction of glomerular podocytes growth and differentiation, thickening of the glomerular basement membrane (GBM), mesangial matrix expansion, focal degeneration of mesangial cells and the mesangial matrix (mesangiolysis), glomerulosclerosis, and tubulointerstitial fibrosis. In Figure 2, morphological and functional alterations to renal glomeruli are indicated as the hallmarks of DKD [3].

Figure 2. Glomerulopathy in diabetes.
Key Cell Models Utilized in DKD Studies
Research has demonstrated that diabetes induces a loss of intrinsic insulin signalling responsiveness in kidney cells. Glomerular podocytes, mesangial cells, glomerular endothelial cells and proximal tubular cells constitute key renal cell populations responsive to insulin signaling. Disruption of insulin signalling pathways in these cell types significantly impairs renal function across various stages of DKD pathogenesis. Moreover, diminished cellular insulin signalling contributes directly to kidney injury and disrupts systemic glucose homoeostasis, thereby exacerbating diabetic pathophysiology.
Very recently, Coward et al. have employed these cellular models under both insulin sensitive and insulin resistant conditions, conducting simultaneous transcriptomic and proteomic profiling for integrated multi-omics analysis. In this study, several consistent changes of individual genes, proteins, and molecular pathways occurring across all insulin resistant kidney cell types, together with cell-line-specific changes occurring in response to insulin resistance, which are replicated in DKD biopsies have been identified. This comprehensive study demonstrates the utility of these four cellular models for mechanistic studies of kidney cell dysfunction in DKD, and establishes a robust data resource to guide future investigations into elucidating underlying kidney signalling pathways and identifying novel therapeutic targets for DKD [4].
Podocytes
Podocytes constitute highly specialized epithelial cells situated along the outer aspect of the glomerular capillary loops, forming an integral component of the glomerular filtration barrier. These cells exhibit a unique structure with foot-like extensions called pedicels that interdigitate and create filtration slits. Podocytes perform an essential function in the selective permeability of the filtration barrier, effectively preventing the pathological leakage of plasma proteins, notably albumin, into the urinary space. Furthermore, in concert with glomerular endothelial cells, podocytes are indispensable for the structural and functional integrity of the GBM, maintenance of its charge-selective properties, and the overall stability of the glomerular capillary tuft. Collectively, these vital roles are profoundly compromised within the diabetic glomerulus [5].
Within the spectrum of early-stage DKD alterations, podocyte dysfunction constitutes a principal pathophysiological event. This dysfunction encompasses a constellation of detrimental changes, including cytoskeletal rearrangement, de-differentiation, apoptosis and dysregulated autophagy. These processes manifest morphologically as foot process effacement (widening, retraction and flattening), reduced cellular motility, aberrant formation of intercellular tight junctions, and a reduction in slit diaphragm length. Concomitant phenomena include glomerular hypertrophy, podocyte detachment and ultimately, podocyte depletion (dropout) from the glomerular tuft [6].
In vitro podocyte experimental models have been successfully employed to recapitulate diabetes-associated phenotypes under diverse pathological conditions, such as cellular injury, chronic hyperglycaemia, and oxidative stress. Consequently, the dysregulation of podocyte growth, differentiation and survival is widely regarded as central to the pathogenesis and progression of DKD [3].
Glomerular endothelial cells
Glomerular endothelial cells form a continuous monolayer lining the luminal surface of glomerular capillaries, representing the cellular interface directly exposed to circulating blood. These cells exhibit a distinctive morphological feature, transcellular pores known as fenestrae, which are critical for the initial phase of plasma ultrafiltration. At their adluminal surface, they are covered by an endothelial glycocalyx layer that occupies the fenestral openings. Functionally, glomerular endothelial cells are essential for modulating glomerular blood flow, preventing intravascular thrombosis, and selectively regulating the passage of solutes and fluids into the nascent urinary filtrate.
Collectively, glomerular endothelial cells, podocytes and GBM constitute the tripartite glomerular filtration barrier. Positioned as the primary vascular barrier, these cells are acutely susceptible to hyperglycemic insult. Pathological alterations in GEnCs, including glycocalyx degradation, loss of fenestral integrity, and endothelial activation, manifest during the early stages of DKD pathogenesis [7]. Nevertheless, the intricate molecular pathways orchestrating hyperglycemia-induced endothelial injury and dysfunction within the glomerulus remain incompletely characterized.
Emerging research is increasingly focused on elucidating the complex intercellular crosstalk involving glomerular endothelial cells, aiming to delineate the specific molecules and signaling cascades implicated. For instance, recent investigations demonstrate that paracrine signaling between glomerular endothelial cells, podocytes, and mesangial cells is significantly mediated by Transforming Growth Factor-beta (TGF-β). This cytokine plays a pivotal role in orchestrating pathogenic responses, contributing directly to the development of glomerular fibrosis observed in DKD patients, thereby underscoring its significance as a key mediator in disease progression [8].
Mesangial cells
Glomerular mesangial cells constitute intrinsic cellular components situated within the glomerular mesangium, anatomically positioned between the afferent and efferent arterioles. These specialized contractile cells perform critical functions in modulating glomerular hemodynamics, specifically through the regulation of capillary luminal diameter to influence intraglomerular blood flow and filtration rate. Functionally, they provide structural support to the glomerular capillary loops and actively secrete a diverse array of bioactive molecules, including extracellular matrix (ECM) components, transforming growth factor-beta (TGF-β), and renin, thereby contributing significantly to the maintenance of renal homeostasis.
Under diabetic conditions, mesangial cells undergo substantial pathological alterations, characterized by cellular proliferation, hypertrophy, and heightened synthesis of matrix proteins. These aberrant changes culminate in distinctive structural hallmarks of diabetic glomerulopathy, notably an increase in the fractional volume of the glomerulus occupied by the mesangium (mesangial expansion), focal degeneration of mesangial cells and the mesangial matrix (mesangiolysis), and the progressive development of glomerulosclerosis. Critically, mesangial matrix expansion exhibits a robust correlation with the clinical progression of DKD.
Notably, mesangial matrix expansion in DKD is not solely a direct consequence of hyperglycemic exposure on mesangial cells. Instead, it is predominantly mediated through intricate intercellular crosstalk involving podocytes, endothelial cells, and inflammatory cells. For instance, emerging evidence indicates that bidirectional signaling between mesangial cells and podocytes, occurring at both early and advanced stages of DKD, is predominantly mediated by vascular endothelial growth factor (VEGF) [8].
Proximal Tubular epithelial cells
The primary physiological functions of renal tubules encompass reabsorption and secretion. Proximal tubular epithelial cells play a central role in reabsorptive processes, recovering approximately two-thirds of the glomerular filtrate and recovering numerous essential solutes from the urinary fluid. These cells mediate the reabsorption of filtered glucose predominantly through sodium-glucose cotransporters 1 and 2 (SGLT1 and SGLT2). Beyond their reabsorptive function, proximal tubular epithelial cells exhibit a potent secretory capacity, particularly under pathological activation. Through mechanisms including chemotaxis, antigen presentation, and autocrine or paracrine cytokine signaling, activated PTECs contribute substantially to the development of tubulointerstitial lesions, renal functional decline, interstitial inflammation, and fibrosis. These processes are intimately associated with the progression of DKD.
Although historical research emphasis has been placed on glomerular injury, accumulating evidence now underscores the critical contribution of renal tubular damage to the pathogenesis of DKD. Multiple pathogenic factors such as hyperglycemia, lipid accumulation, oxidative stress, hypoxia, renin-angiotensin-aldosterone system (RAAS) activation, endoplasmic reticulum (ER) stress, inflammatory responses, epithelial-mesenchymal transition (EMT), and programmed cell death have been demonstrated to induce tubular injury and accelerate the progression of DKD [9].
Emerging studies indicate that proximal tubular epithelial cells engage in critical cellular crosstalk with other key renal cell types, including podocytes, endothelial cells, mesangial cells, fibroblasts, and monocytes. Through these interactions, proximal tubular epithelial cells actively promote disease advancement, and are thereby established as a promising therapeutic target in DKD. A deeper understanding of proximal tubular epithelial cell-targeted interventions and the development of precise targeting strategies may thus pave the way for novel treatment modalities against DKD [8, 10].
Conclusion
Integrated research efforts focusing on intercellular communication and targeted interventions against specific renal cell dysfunctions will be essential for developing effective strategies to halt or reverse the progression of DKD. Key kidney cell types—podocytes, glomerular endothelial cells, mesangial cells and proximal tubular epithelial cells—are critically implicated in the pathogenesis of DKD. These cells provide reproducible and cost-effective in vitro models for investigating the signalling pathways underlying DKD progression. Such cellular systems are indispensable for elucidating molecular mechanisms, validating novel biomarkers, and screening potential therapeutic agents for DKD intervention.
References
[1] S. Saber, R. Abdelhady, M.A. Elhemely, E.A. Elmorsy, R.S. Hamad, M.A. Abdel-Reheim, A.F. El-Kott, M.A. AlShehri, K. Morsy, S. Negm, A.Y. Kira, Nanoscale Systems for Local Activation of Hypoxia-Inducible Factor-1 Alpha: A New Approach in Diabetic Wound Management, Int J Nanomedicine 19 (2024) 13735-13762.
[2] S. Gupta, M. Dominguez, L. Golestaneh, Diabetic Kidney Disease: An Update, The Medical clinics of North America 107(4) (2023) 689-705.
[3] M.C. Thomas, M. Brownlee, K. Susztak, K. Sharma, K.A. Jandeleit-Dahm, S. Zoungas, P. Rossing, P.H. Groop, M.E. Cooper, Diabetic kidney disease, Nature reviews. Disease primers 1 (2015) 15018.
[4] A.C. Lay, V.D.T. Tran, V. Nair, V. Betin, J.A. Hurcombe, A.F. Barrington, R.J. Pope, F. Burdet, F. Mehl, D. Kryvokhyzha, A. Ahmad, M.C. Sinton, P. Lewis, M.C. Wilson, R. Menon, E. Otto, K.J. Heesom, M. Ibberson, H.C. Looker, R.G. Nelson, W. Ju, M. Kretzler, S.C. Satchell, M.F. Gomez, R.J.M. Coward, Profiling of insulin-resistant kidney models and human biopsies reveals common and cell-type-specific mechanisms underpinning Diabetic Kidney Disease, Nature communications 15(1) (2024) 10018.
[5] J. Reiser, M.M. Altintas, Podocytes, F1000Research 5 (2016).
[6] S. Husain, Role of Podocyte in Kidney Disease, Frontiers in bioscience (Landmark edition) 29(7) (2024) 250.
[7] X. Fan, M. Yang, Y. Lang, S. Lu, Z. Kong, Y. Gao, N. Shen, D. Zhang, Z. Lv, Mitochondrial metabolic reprogramming in diabetic kidney disease, Cell death & disease 15(6) (2024) 442.
[8] S. Jiang, M. Luo, X. Bai, P. Nie, Y. Zhu, H. Cai, B. Li, P. Luo, Cellular crosstalk of glomerular endothelial cells and podocytes in diabetic kidney disease, Journal of cell communication and signaling 16(3) (2022) 313-331.
[9] Y. Wang, M. Jin, C.K. Cheng, Q. Li, Tubular injury in diabetic kidney disease: molecular mechanisms and potential therapeutic perspectives, Frontiers in endocrinology 14 (2023) 1238927.
[10] H. Li, W. Dai, Z. Liu, L. He, Renal Proximal Tubular Cells: A New Site for Targeted Delivery Therapy of Diabetic Kidney Disease, Pharmaceuticals 15(12) (2022) 1494.
Copyright - Unless otherwise stated all contents of this website are AcceGen™ All Rights Reserved – Full details of the use of materials on this site please refer to AcceGen Editorial Policy – Guest Posts are welcome, by submitting a guest post to AcceGen you are agree to the AcceGen Guest Post Agreement – Any concerns please contact marketing@accegen.com